
Introduction: Navigating Medical Injection Molding Compliance
Medical injection molding ranks among the most tightly regulated manufacturing processes in the industry. A single compliance oversight—whether in material traceability, process validation, or documentation—can delay product launches by months or trigger costly recalls that damage both finances and reputation.
Medical device manufacturers face a daunting web of FDA regulations, ISO standards, and quality management requirements. Understanding FDA 21 CFR Part 820, achieving ISO 13485 certification, navigating biocompatibility testing, and maintaining audit-ready documentation creates a complex compliance burden.
Without expert guidance, managing these overlapping requirements can feel overwhelming.
This guide explains the regulatory landscape for medical injection molding. We'll break down FDA 21 CFR Part 820 requirements, ISO 13485 certification essentials, material biocompatibility standards (USP Class VI vs. ISO 10993), and actionable compliance steps from design through production.
Key Takeaways
- FDA's new QMSR (Feb 2026) harmonizes U.S. regulations with ISO 13485:2016 globally
- Class II recalls from manufacturing defects link directly to inadequate process validation
- Process validation (IQ/OQ/PQ) is mandatory for injection molding where inspection alone can't verify quality
- Material selection requires ISO 10993-1 risk-based biocompatibility assessment, not just USP Class VI
- Manufacturing partners with ISO 13485 experience prevent costly downstream regulatory issues
Understanding Medical Injection Molding Regulatory Landscape
Why Medical Device Manufacturing Requires Heightened Regulatory Scrutiny
Medical devices directly impact patient safety, making regulatory compliance absolutely non-negotiable. Unlike commercial plastic products, medical components must meet stringent quality standards because failures can result in serious injury or death.
The FDA uses a risk-based classification system that determines the level of regulatory control required:
- Class I (Low Risk): Basic instruments with minimal patient risk, subject to General Controls (registration, labeling, adulteration prevention). Most are exempt from 510(k) premarket notification, though design controls still apply to specific devices like surgeon's gloves
- Class II (Moderate Risk): Devices requiring Special Controls beyond general requirements, typically needing 510(k) clearance and full compliance with 21 CFR 820 design controls
- Class III (High Risk): Life-supporting or life-sustaining devices requiring Premarket Approval (PMA) and the strictest regulatory oversight

Recent data reveals a concerning upward trend: medical device recalls increased from 33 in 2020 to 61 in 2023, with Class II recalls consistently outnumbering Class I and III combined.
Manufacturing defects—including cracked components, device separations during use, and structural failures—represent significant root causes, directly linked to inadequate process validation and quality control in injection molding operations.
Key Regulatory Bodies and Their Roles
The FDA (United States) controls premarket approval pathways (510(k), PMA) and enforces Quality System Regulations through facility inspections. The FDA's new Quality Management System Regulation (QMSR), effective February 2, 2026, marks a significant shift by incorporating ISO 13485:2016 by reference.
International regulatory bodies including the European Union (MDR/IVDR), Health Canada, and Australia's TGA all reference ISO 13485 as their quality management framework. This global harmonization allows manufacturers to use a single QMS structure to satisfy multiple jurisdictions, provided they also meet specific local requirements.
ISO standards serve as the global framework that regulatory bodies reference. ISO 13485 (quality management), ISO 14971 (risk management), and ISO 10993 (biocompatibility) form the foundation of international medical device compliance.
The Compliance Journey: From Design to Market
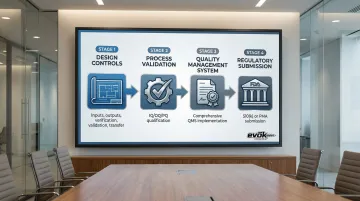
Understanding these regulatory bodies sets the stage for navigating the actual compliance process. Regulatory compliance starts at the design phase, not after manufacturing begins. The typical pathway follows this sequence:
- Design Controls → Establish design inputs, outputs, verification, validation, and transfer procedures
- Process Validation → Complete IQ/OQ/PQ qualification for manufacturing processes
- Quality Management System → Implement comprehensive QMS covering all operations
- Regulatory Submission → Prepare and submit 510(k) or PMA with supporting documentation
Critical Documentation Requirements
Two documentation systems anchor this compliance journey:
- Design History File (DHF): Compilation of records describing the design history and demonstrating that development followed the approved design plan
- Device Master Record (DMR): Compilation of procedures and specifications serving as the "recipe" for production, including drawings, formulations, and component specifications
Confusion between these files is a common compliance pitfall during FDA inspections.
FDA Requirements for Medical Device Manufacturing
FDA 21 CFR Part 820: Quality System Regulation (QSR)
Medical device manufacturers face mounting pressure to navigate complex, evolving FDA regulations while maintaining production efficiency. The Quality Management System Regulation (QMSR) amends 21 CFR Part 820 to incorporate ISO 13485:2016 by reference, harmonizing U.S. requirements with global standards.
This regulation establishes current Good Manufacturing Practices (cGMP) for medical devices.
Key coverage areas:
- Design controls and design transfer
- Document and change controls
- Purchasing controls and supplier management
- Production and process controls
- Corrective and Preventive Action (CAPA)
- Management responsibility and resource allocation
For injection molding operations specifically, 21 CFR Part 820 mandates:
- Complete process validation with statistical justification
- Equipment qualification (IQ/OQ/PQ)
- Environmental controls appropriate to device classification
- Comprehensive documentation at every production stage
The QMSR retains specific FDA requirements for labeling/packaging controls (820.45) and records control (820.35). These ensure consistency with the Federal Food, Drug, and Cosmetic Act.
Design Controls and Design for Manufacturability (DFM)
FDA design controls (21 CFR 820.30) are mandatory for all Class II/III devices and select Class I devices. The six essential elements are:
- Design Input: Physical and performance characteristics derived from user needs
- Design Output: Device specifications, drawings, and documentation allowing evaluation against inputs
- Design Verification: Objective evidence (test reports) confirming Output meets Input
- Design Validation: Objective evidence that specifications conform to User Needs under actual/simulated conditions
- Design Transfer: Procedures ensuring design is correctly translated into production specifications
- Design Changes: Identification, documentation, validation, and approval before implementation
These controls connect directly to Design for Manufacturability (DFM). DFM considerations for medical devices must account for regulatory requirements from the start—including material traceability systems, process capability analysis, and validation requirements.
Design Failure Mode and Effects Analysis (DFMEA) identifies potential compliance risks early when modifications are most cost-effective. Manufacturing partners with deep DFM expertise—like EVOK with 25 years of injection molding experience and Six Sigma capabilities—help identify and mitigate compliance risks during the design phase.
This prevents costly downstream regulatory issues.
Process Validation Requirements
Process validation is required when the results of a process cannot be fully verified by subsequent inspection and testing.
For injection molding, internal stress distribution and part strength often cannot be non-destructively tested, making validation mandatory.
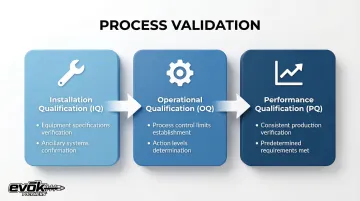
The three stages:
| Stage | Definition & Requirements |
|---|---|
| Installation Qualification (IQ) | Establish by objective evidence that all key aspects of process equipment and ancillary systems adhere to approved specifications |
| Operational Qualification (OQ) | Establish process control limits and action levels that result in product meeting all predetermined requirements |
| Performance Qualification (PQ) | Establish that the process, under anticipated conditions, consistently produces product meeting all predetermined requirements |
For injection molding specifically, validation must cover:
- Temperature profiles across all zones
- Pressure curves (injection, holding, back pressure)
- Cycle times and cooling rates
- Dimensional tolerances and critical features
- Material flow characteristics and knit line locations
Documentation requirements:
- Detailed validation protocols approved before execution
- Test results with statistical analysis
- Sampling plans based on valid statistical rationale
- Ongoing process monitoring procedures (Statistical Process Control)
FDA inspections frequently cite inadequate statistical justification for sampling plans as a common 483 observation. Testing only 3 units for shelf-life without documented rationale is a typical example.
Material Traceability and Documentation
Complete traceability from raw material lot to finished device is non-negotiable. For each production lot, the Device History Record (DHR) must document:
- Dates of manufacture
- Quantity manufactured and released for distribution
- Acceptance records demonstrating DMR compliance
- Labeling used for each production unit/lot
- Unique Device Identifier (UDI) or control number
Required supplier documentation:
- Certificate of Compliance (CoC): Verifies material conforms to specifications
- Certificate of Analysis (CoA): Provides test data for specific material lots
- Biocompatibility test reports: ISO 10993 endpoint data
- Material Safety Data Sheets (MSDS): Chemical safety information
Change control is critical. When material suppliers make formulation changes, manufacturers must document the change, assess its impact, and potentially re-validate affected processes.
FDA Inspections and Audit Readiness
During FDA inspections of medical device manufacturing facilities, investigators examine:
- Design History Files and Device Master Records
- Process validation documentation (IQ/OQ/PQ protocols and results)
- CAPA effectiveness and trending
- Complaint handling and adverse event reporting
- Supplier controls and incoming inspection records
Common 483 observations related to injection molding:
- Inadequate process validation or lack of statistical justification
- Insufficient documentation of process parameters
- Failure to validate processes where results cannot be fully verified
- Ineffective CAPA systems that don't prevent recurrence
- Missing or incomplete DHR documentation
Maintaining audit readiness:
Rather than scrambling before inspections, implement continuous audit readiness. This means daily adherence to QMS procedures, regular internal audits, management review meetings, and proactive CAPA trending analysis.
Treat every production run as if an inspector will review the documentation tomorrow.
ISO 13485 Certification: What Manufacturers Need to Know
ISO 13485:2016 Overview and Requirements
ISO 13485:2016 is the international standard for quality management systems specific to medical devices. It follows ISO 9001's framework but includes critical medical device requirements:
- Management Responsibility: Top management commitment, customer focus, quality policy, and planning
- Resource Management: Provision of resources, human resources, infrastructure, and work environment
- Product Realization: Planning, customer-related processes, design/development, purchasing, production/service provision
- Measurement, Analysis, and Improvement: Monitoring, control of non-conforming product, data analysis, and CAPA
How ISO 13485 differs from ISO 9001:
- More prescriptive documentation and record requirements
- Requires integration of risk management (ISO 14971) into product realization
- Prioritizes regulatory compliance and safety over continuous improvement
- Built specifically for regulatory purposes
Risk Management and ISO 14971
ISO 13485 requires a risk management process throughout the product lifecycle. ISO 14971:2019 provides the framework for identifying hazards, estimating and evaluating risks, controlling risks, and monitoring effectiveness.
Application to injection molding:
- Process risk identification: Short shots, flash, sink marks, warpage, contamination, dimensional variation
- Risk controls: Process parameters, environmental controls, inspection protocols, operator training
- Effectiveness monitoring: Statistical Process Control (SPC), ongoing validation, trend analysis
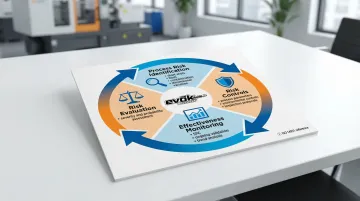
Risk management isn't a one-time activity. It's an ongoing process integrated into design, production, and post-market surveillance.
Clean Room and Environmental Controls
ISO 14644-1:2015 specifies classification of air cleanliness by particle concentration. Clean rooms are required when contamination control is necessary to meet device specifications—particularly for sterile barriers, implantable devices, or particulate-sensitive components.
Classification system: Based on concentration of airborne particles (0.1 µm to 5 µm) measured using light scattering particle counters. ISO Class 5 (formerly Class 100) through ISO Class 9 (formerly Class 100,000) define progressively less stringent cleanliness levels.
Environmental monitoring requirements:
- Particulate counts at defined intervals
- Microbial monitoring (surface and air sampling)
- Temperature and humidity control with continuous recording
- Differential pressure monitoring between zones
Clean room manufacturing involves significantly higher costs and operational complexity than standard environments.
Classification should be based on actual device requirements, not arbitrary standards.
Documentation and Record Keeping
ISO 13485 requires a tiered documentation system:
- Quality Manual: Top-level QMS description and scope
- Procedures (SOPs): Detailed process descriptions
- Work Instructions: Step-by-step operator guidance
- Records: Objective evidence of activities performed
Document control requirements:
- Revision management with change history
- Approval processes before implementation
- Distribution control ensuring current versions are used
- Periodic review and updates
Record retention: Records must be retained for a period equivalent to the design and expected life of the device, but no less than 2 years from commercial release.
Electronic records: 21 CFR Part 11 applies to electronic records and signatures, requiring controls for security, integrity, and attribution.
Supplier Management and Incoming Material Controls
ISO 13485 requires rigorous supplier qualification and ongoing monitoring:
- Supplier qualification: Assessment of capability to meet requirements before approval
- Incoming inspection: Verification that materials and components conform to specifications
- Supplier audits: Periodic on-site audits for critical materials and processes
- Performance monitoring: Trending of quality metrics, on-time delivery, and non-conformances
For medical-grade materials, incoming inspection must verify:
- Material certification documents (CoC/CoA)
- Lot traceability information
- Biocompatibility test reports
- Physical and chemical properties
Supplier change control is critical—manufacturers must be notified of any formulation changes and assess potential impact on device performance and biocompatibility.
Material Selection and Biocompatibility Standards
Medical-Grade Materials: USP Class VI and ISO 10993
Understanding the difference between historical and current biocompatibility standards is essential:
USP Class VI: A set of in vivo biological reactivity tests (USP <88>) historically used as a baseline. While still referenced, FDA guidance emphasizes that USP Class VI alone is often insufficient for modern biocompatibility evaluation.
ISO 10993-1: The current global standard for biological evaluation within a risk management process. It uses a matrix based on the nature and duration of body contact to determine relevant endpoints.
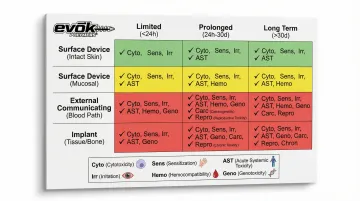
The ISO 10993 test matrix:
| Nature of Contact | Duration | Required Endpoints (Examples) |
|---|---|---|
| Surface Device (Intact Skin) | Limited (<24h) | Cytotoxicity, Sensitization, Irritation |
| Surface Device (Mucosal Membrane) | Prolonged (24h-30d) | Above + Acute Systemic Toxicity, Subacute Toxicity, Implantation |
| External Communicating (Blood Path, Indirect) | Limited (<24h) | Cytotoxicity, Sensitization, Irritation, Acute Systemic Toxicity, Hemocompatibility |
| Implant (Tissue/Bone) | Long Term (>30d) | All above + Genotoxicity, Chronic Toxicity, Carcinogenicity |
Critical clarification: Material suppliers provide biocompatibility data, but device manufacturers are responsible for the final biocompatibility assessment based on actual device configuration, contact type, and duration.
Common Medical-Grade Thermoplastics and Elastomers
The most common materials for medical injection molding each offer distinct advantages:
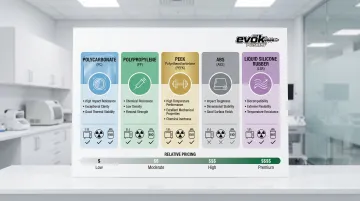
Polycarbonate (PC) dominates applications requiring optical clarity—housings, connectors, and transparent components. It withstands gamma and ethylene oxide (EtO) sterilization while providing high impact strength and heat resistance.
Polypropylene (PP) remains the workhorse for syringes, containers, and labware. Its chemical resistance and low cost make it economical, plus it tolerates steam autoclave sterilization—a significant advantage for reusable devices.
For demanding structural applications, PEEK (Polyetheretherketone) delivers exceptional strength and heat resistance. Surgical instruments and implants benefit from its biocompatibility and ability to withstand all major sterilization methods.
ABS (Acrylonitrile-Butadiene-Styrene) suits non-critical housings where impact resistance matters but body contact is minimal. It processes easily and tolerates EtO and low-temperature sterilization methods.
Liquid Silicone Rubber (LSR) serves soft-touch applications—seals, gaskets, and implantable devices requiring flexibility. Its biocompatibility profile and temperature resistance make it compatible with steam autoclave, gamma, and EtO sterilization.
Medical-grade materials carry a 20-40% cost premium over commercial-grade plastics due to controlled manufacturing, batch testing, and complete documentation needs.
Material Certification and Traceability Requirements
Selecting the right material is only the first step. Regulatory compliance demands rigorous documentation for every shipment:
- Certificate of Compliance (CoC): Confirms material meets specified requirements
- Certificate of Analysis (CoA): Provides test results for the specific lot
- Biocompatibility test reports: ISO 10993 endpoint data or master file references
- Material Safety Data Sheets (MSDS/SDS): Chemical composition and safety information
Lot traceability throughout the supply chain is mandatory. Manufacturers must maintain records linking:
- Raw material lot numbers to incoming inspection records
- Material lots to production runs (via DHR)
- Production runs to finished device serial numbers or lot codes
- Finished devices to distribution records
Change control for material formulations: When suppliers modify formulations—even minor changes—manufacturers must:
- Receive formal change notification
- Assess impact on device performance and biocompatibility
- Determine if re-testing or re-validation is required
- Document the change control decision and rationale
- Update Device Master File if necessary
Selecting a Compliant Manufacturing Partner
Essential Certifications and Capabilities
When evaluating injection molding partners for medical device manufacturing, certain certifications and capabilities are essential:
Must-have certifications:
- ISO 13485 certification: Demonstrates conformance to medical device quality management requirements
- FDA registration: Required if manufacturing finished devices for U.S. market
- Clean room capabilities: ISO 14644 classification appropriate to your device (if required)
Critical capabilities:
- Process validation expertise: Documented experience with IQ/OQ/PQ protocols and statistical validation
- Design support and DFM: Ability to optimize designs for regulatory compliance from the start
- Material expertise: Knowledge of medical-grade materials, biocompatibility requirements, and sterilization compatibility
Companies like EVOK, with 25 years of injection molding experience and Six Sigma expertise, can help identify and mitigate compliance risks during the design phase—preventing costly downstream regulatory issues.
Early DFM engagement ensures that material traceability systems, process capability requirements, and validation needs are built into the design rather than retrofitted later.
Questions to Ask Potential Manufacturing Partners
Regulatory experience:
- What is your experience with FDA-regulated devices in our classification?
- Can you provide references from medical device clients in similar applications?
- How many FDA inspections have you undergone, and what were the results?
Quality systems:
- Describe your CAPA process and provide examples of effectiveness.
- How do you handle change control for materials, processes, and documentation?
- What is your approach to supplier qualification and ongoing monitoring?
Process capabilities:
- What is your process for IQ/OQ/PQ validation?
- How do you establish and monitor critical process parameters?
- What statistical methods do you use for process capability analysis?
Transparency and partnership:
- Can you provide detailed cost breakdowns and timeline expectations?
- How do you communicate when issues or delays arise?
- What level of design support do you provide before tooling?
Transparency in pricing and timeline expectations is critical. Compliant manufacturing involves more steps, documentation, and validation than commercial production, and your partner should clearly communicate these requirements upfront.
Red Flags and Warning Signs
Once you've asked the right questions, watch for red flags. Avoid manufacturers who:
- Lack documented quality management systems or refuse to share QMS documentation
- Cannot provide validation protocols or examples of completed validations
- Have no experience with FDA inspections or regulatory audits
- Offer significantly lower pricing without explaining what corners are being cut
- Are unwilling to discuss their supplier qualification processes
- Cannot demonstrate material traceability systems
- Lack clean room capabilities when your device classification requires them
The lowest-cost provider is seldom the best choice for medical device manufacturing. Compliance shortcuts always cost more in the long run through:
- Product launch delays and expensive rework to address regulatory deficiencies
- Failed FDA inspections and subsequent corrective actions
- Product recalls with lasting damage to company reputation
Invest in a qualified partner from the start. The premium paid for documented compliance capabilities is insignificant compared to the cost of regulatory failures.
Frequently Asked Questions
What is medical injection molding?
Medical injection molding creates plastic components and devices for healthcare applications under strict quality controls. The process requires comprehensive documentation, process validation, and material traceability that exceed commercial manufacturing standards to ensure patient safety.
What is FDA 21 CFR Part 820 for medical devices?
Part 820 establishes the FDA's Quality System Regulation (QSR) with current Good Manufacturing Practices for medical device manufacturers. The new QMSR (effective February 2026) incorporates ISO 13485:2016 by reference, harmonizing U.S. requirements with global standards.
Is ISO 13485 mandatory for medical devices?
ISO 13485 certification isn't legally required by the FDA but serves as the international standard for medical device quality management. International markets (EU, Canada, Australia) often require it, and the FDA's new QMSR incorporates it by reference, making it the de facto U.S. standard.
What ISO standards apply to medical injection molding?
Key standards include ISO 13485 (quality management), ISO 14971 (risk management), ISO 10993 (biocompatibility), ISO 14644 (clean rooms), ISO 11607 (sterilized device packaging), and ISO 20457 (plastic part tolerances). Together they create a comprehensive compliance framework.
What is the difference between ISO 13485 and the EU MDR?
ISO 13485 defines quality management system requirements for manufacturers, while EU MDR is the legal regulatory framework for placing devices on the European market. ISO 13485 certification supports MDR compliance but doesn't automatically guarantee it.
What are the material requirements for plastics used in medical injection molding?
Plastics must be medical-grade with biocompatibility testing per ISO 10993-1 and complete documentation (CoC, CoA, biocompatibility reports, MSDS). They require full lot traceability and compatibility with required sterilization methods without degradation.
What tolerances are typical for medical plastic injection molding?
Typical tolerances range from ±0.001" to ±0.005" depending on the application, tighter than commercial plastics. Critical dimensions follow ISO 20457 and ASME Y14.5 (GD&T) with statistical process control (Cpk ≥1.33) validation.


